
Elektroniczna struktura pasmowa - Electronic band structure
W fizyce półprzewodnikowym The struktura elektroniczny zespół (lub po prostu struktura pasma ) substancji stałej opisuje zakres energii poziomu, że elektrony mogą być w nim, jak również zakresy energii, która nie może być (zwane pasma wzbronionego lub zabronione zespoły ).
Teoria pasmowa wyprowadza te pasma i przerwy wzbronione poprzez badanie dozwolonych funkcji fal kwantowo-mechanicznych dla elektronu w dużej, okresowej sieci atomów lub cząsteczek. Teoria pasmowa została z powodzeniem wykorzystana do wyjaśnienia wielu właściwości fizycznych ciał stałych, takich jak oporność elektryczna i absorpcja optyczna , i stanowi podstawę zrozumienia wszystkich urządzeń półprzewodnikowych (tranzystory, ogniwa słoneczne itp.).
Dlaczego występują pasma i przerwy między pasmami?

Elektrony pojedynczego, izolowanego atomu zajmują orbitale atomowe, z których każdy ma dyskretny poziom energii . Kiedy dwa lub więcej atomów łączy się, tworząc cząsteczkę , ich orbitale atomowe nakładają się. W Zakaz Pauliego mówi, że żadne dwa elektrony mogą mieć te same liczby kwantowe w cząsteczce. Jeśli więc dwa identyczne atomy łączą się, tworząc cząsteczkę dwuatomową , każdy orbital atomowy dzieli się na dwa orbitale molekularne o różnej energii, umożliwiając elektronom z poprzednich orbitali atomowych zajęcie nowej struktury orbitalnej bez posiadania takiej samej energii.
Podobnie, jeśli duża liczba N identycznych atomów łączy się, tworząc ciało stałe, takie jak sieć krystaliczna , orbitale atomowe atomów nakładają się. Ponieważ zasada wykluczenia Pauliego mówi, że żadne dwa elektrony w ciele stałym nie mają takich samych liczb kwantowych, każdy orbital atomowy dzieli się na N dyskretnych orbitali molekularnych, każdy o innej energii. Ponieważ liczba atomów w makroskopowym kawałku ciała stałego jest bardzo duża (N~10 22 ), liczba orbitali jest bardzo duża, a zatem są one bardzo blisko siebie rozmieszczone pod względem energii (rzędu 10 -22 eV). Energia sąsiednich poziomów jest tak blisko siebie, że można je uznać za kontinuum, pasmo energii.
To tworzenie pasm jest głównie cechą najbardziej zewnętrznych elektronów (elektronów walencyjnych ) w atomie, które są zaangażowane w wiązania chemiczne i przewodnictwo elektryczne . Wewnętrzne orbitale elektronowe nie nakładają się w znacznym stopniu, więc ich pasma są bardzo wąskie.
Pasma wzbronione są zasadniczo pozostałymi zakresami energii niepokrytymi przez żadne pasmo, w wyniku skończonych szerokości pasm energii. Pasma mają różne szerokości, przy czym szerokości zależą od stopnia nakładania się orbitali atomowych, z których powstają. Dwa sąsiednie pasma mogą po prostu nie być wystarczająco szerokie, aby w pełni pokryć zakres energii. Na przykład pasma związane z orbitalami rdzenia (takie jak elektrony 1s ) są niezwykle wąskie ze względu na niewielkie nakładanie się sąsiednich atomów. W rezultacie między pasmami rdzenia występują zwykle duże przerwy wzbronione. Wyższe pasma wiążą się ze stosunkowo większymi orbitalami z większym nakładaniem się, stając się coraz szerszymi przy wyższych energiach, tak że nie ma przerw wzbronionych przy wyższych energiach.
Podstawowe koncepcje
Założenia i granice teorii struktury pasmowej
Teoria pasmowa jest tylko przybliżeniem do stanu kwantowego ciała stałego, który dotyczy ciał stałych składających się z wielu identycznych atomów lub cząsteczek połączonych ze sobą. Oto założenia niezbędne, aby teoria pasmowa była słuszna:
- System o nieskończonej wielkości : aby pasma były ciągłe, kawałek materiału musi składać się z dużej liczby atomów. Ponieważ makroskopowy kawałek materiału zawiera około 10 22 atomów, nie jest to poważne ograniczenie; teoria pasmowa odnosi się nawet do mikroskopijnych rozmiarów tranzystorów w układach scalonych . Dzięki modyfikacjom pojęcie struktury pasmowej można również rozszerzyć na układy, które są „duże” tylko w pewnych wymiarach, takie jak dwuwymiarowe układy elektronowe .
- Układ jednorodny : Struktura pasmowa jest nieodłączną właściwością materiału, która zakłada, że materiał jest jednorodny. W praktyce oznacza to, że skład chemiczny materiału musi być jednolity w całym kawałku.
- Brak interaktywności : Struktura pasmowa opisuje „stany jednoelektronowe”. Istnienie tych stanów zakłada, że elektrony poruszają się w potencjale statycznym bez dynamicznego oddziaływania z drganiami sieci , innymi elektronami, fotonami itp.
Powyższe założenia są łamane w wielu ważnych sytuacjach praktycznych, a wykorzystanie struktury pasmowej wymaga ścisłego sprawdzania ograniczeń teorii pasmowej:
- Niejednorodności i interfejsy: W pobliżu powierzchni, połączeń i innych niejednorodności struktura pasmowa jest zaburzona. Występują nie tylko lokalne zakłócenia na małą skalę (np. stany powierzchniowe lub stany domieszki wewnątrz pasma wzbronionego), ale także lokalne nierównowagi ładunków. Te nierównowagi ładunków mają efekty elektrostatyczne, które rozciągają się głęboko na półprzewodniki, izolatory i próżnię (patrz domieszkowanie , zginanie pasma ).
- Wzdłuż tej samej linii, w większości efekty elektroniczne ( pojemności , przewodnictwa elektrycznego , polem elektrycznym przesiewania ) obejmują fizyki elektronów przechodzących przez powierzchnie i / lub w pobliżu interfejsów. Pełny opis tych efektów, w postaci obrazu struktury pasmowej, wymaga przynajmniej szczątkowego modelu oddziaływań elektron-elektron (patrz ładunek kosmiczny , zginanie pasma ).
- Małe systemy: W przypadku systemów, które są małe w każdym wymiarze (np. mała cząsteczka lub kropka kwantowa ), nie ma ciągłej struktury pasmowej. Przejście między małymi i dużymi wymiarami to królestwo fizyki mezoskopowej .
- Materiały silnie skorelowane (np. izolatory Motta ) po prostu nie mogą być rozumiane w kategoriach stanów jednoelektronowych. Struktury pasm elektronicznych tych materiałów są słabo zdefiniowane (lub przynajmniej nie są jednoznacznie określone) i mogą nie dostarczać użytecznych informacji o ich stanie fizycznym.
Symetria krystaliczna i wektory falowe
.svg)

Obliczenia struktury pasmowej wykorzystują okresowość sieci krystalicznej, wykorzystując jej symetrię. Pojedyncza elektronów równanie Schrödingera jest rozwiązane za elektronu w potencjale kraty okresowego, dając elektronów Bloch jako roztwory:
- ,

gdzie k nazywamy wektorem falowym. Dla każdej wartości k istnieje wiele rozwiązań równania Schrödingera oznaczonego przez n , indeks pasma, który po prostu numeruje pasma energii. Każdy z tych poziomów energetycznych ewoluuje płynnie ze zmianami k , tworząc gładkie pasmo stanów. Dla każdego pasma możemy zdefiniować funkcję E n ( k ), która jest relacją dyspersji elektronów w tym paśmie.
Wektor falowy przyjmuje dowolną wartość w strefie Brillouina , która jest wielościanem w przestrzeni falowej ( sieci odwrotnej ), która jest związana z siecią kryształu. Wektory falowe poza strefą Brillouina odpowiadają po prostu stanom, które są fizycznie identyczne ze stanami w strefie Brillouina. Specjalne punkty/linie o wysokiej symetrii w strefie Brillouina mają przypisane etykiety takie jak Γ, Δ, Λ, Σ (patrz rys. 1).
Trudno wyobrazić sobie kształt pasma jako funkcję wektora falowego, ponieważ wymagałoby to wykresu w przestrzeni czterowymiarowej, E vs. k x , k y , k z . W literaturze naukowej często można zobaczyć wykresy struktury pasmowej, które pokazują wartości E n ( k ) dla wartości k wzdłuż linii prostych łączących punkty symetrii, często oznaczanych jako Δ, Λ, Σ lub [100], [111], oraz [110] , odpowiednio. Inną metodą wizualizacji struktury pasmowej jest wykreślenie izopowierzchni o stałej energii w przestrzeni wektora falowego, pokazującej wszystkie stany o energii równej określonej wartości. Izopowierzchnia stanów o energii równej poziomowi Fermiego nazywana jest powierzchnią Fermiego .
Energetyczne przerwy wzbronione można sklasyfikować za pomocą wektorów falowych stanów otaczających przerwę wzbronioną:
- Bezpośrednie pasmo wzbronione : stan o najniższej energii powyżej pasma wzbronionego ma to samo k, co stan o najwyższej energii poniżej pasma wzbronionego.
- Pośrednia przerwa wzbroniona : najbliższe stany powyżej i poniżej pasma wzbronionego nie mają tej samej wartości k .
Asymetria: Struktury pasmowe w niekrystalicznych ciałach stałych
Chociaż elektronowe struktury pasmowe są zwykle związane z materiałami krystalicznymi , quasi-krystaliczne i amorficzne ciała stałe mogą również wykazywać przerwy wzbronione. Są one nieco trudniejsze do zbadania teoretycznie, ponieważ brakuje im prostej symetrii kryształu i zwykle nie jest możliwe określenie dokładnej zależności dyspersyjnej. W rezultacie praktycznie wszystkie istniejące prace teoretyczne nad elektronową strukturą pasmową ciał stałych koncentrowały się na materiałach krystalicznych.
Gęstość stanów
Funkcja gęstości stanów g ( E ) jest zdefiniowana jako liczba stanów elektronowych na jednostkę objętości, na jednostkę energii, dla energii elektronów bliskich E .
Funkcja gęstości stanów jest istotna przy obliczaniu efektów w oparciu o teorię pasmową. W złotej regule Fermiego , obliczaniu szybkości absorpcji optycznej , podaje ona zarówno liczbę elektronów pobudliwych, jak i liczbę stanów końcowych elektronu. Pojawia się w obliczeniach przewodnictwa elektrycznego, gdzie podaje liczbę stanów ruchomych, oraz w obliczaniu szybkości rozpraszania elektronów, gdzie podaje liczbę stanów końcowych po rozproszeniu.
Dla energii wewnątrz przerwy energetycznej g ( E ) = 0.
Wypełnianie opasek

W równowadze termodynamicznej prawdopodobieństwo wypełnienia stanu energii E przez elektron jest określone przez rozkład Fermi-Diraca, rozkład termodynamiczny, który uwzględnia zasadę wykluczenia Pauliego :

gdzie:
- k B T jest iloczynem stałej Boltzmanna i temperatury , a
- µ to całkowity potencjał chemiczny elektronów, czyli poziom Fermiego (w fizyce półprzewodników wielkość ta jest częściej oznaczana E F ). Poziom Fermiego ciała stałego jest bezpośrednio związany z napięciem na tym ciele stałym, mierzonym woltomierzem. Konwencjonalnie, na wykresach struktury pasmowej poziom Fermiego przyjmuje się jako zero energii (wybór arbitralny).
Gęstość elektronów w materiale to po prostu całka rozkładu Fermiego-Diraca razy gęstość stanów:

Chociaż istnieje nieskończona liczba pasm, a zatem nieskończona liczba stanów, w tych pasmach można umieścić tylko skończoną liczbę elektronów. Preferowana wartość liczby elektronów jest konsekwencją elektrostatyki: chociaż powierzchnia materiału może być naładowana, wewnętrzna masa materiału woli być ładowana. Warunek neutralności ładunku oznacza, że N / V musi odpowiadać gęstości protonów w materiale. Aby tak się stało, materiał elektrostatycznie dostosowuje się, przesuwając swoją strukturę pasmową w górę lub w dół pod względem energii (a tym samym przesuwając g ( E )), aż znajdzie się w prawidłowej równowadze w odniesieniu do poziomu Fermiego.
Nazwy pasm w pobliżu poziomu Fermiego (pasmo przewodnictwa, pasmo walencyjne)
Bryła ma nieskończoną liczbę dozwolonych pasm, tak jak atom ma nieskończenie wiele poziomów energii. Jednak większość zespołów ma po prostu zbyt dużą energię i zwykle są lekceważone w zwykłych okolicznościach. I odwrotnie, istnieją bardzo niskie pasma energii związane z orbitalami rdzenia (takie jak elektrony 1s ). Te niskoenergetyczne pasma rdzenia są również zwykle pomijane, ponieważ przez cały czas pozostają wypełnione elektronami, a zatem są obojętne. Podobnie materiały mają kilka przerw wzbronionych w całej swojej strukturze pasmowej.
Najważniejsze pasma i przerwy wzbronione — te istotne dla elektroniki i optoelektroniki — to te o energiach zbliżonych do poziomu Fermiego. Wstęgi i przerwy wstęgowe w pobliżu poziomu Fermiego otrzymują specjalne nazwy, w zależności od materiału:
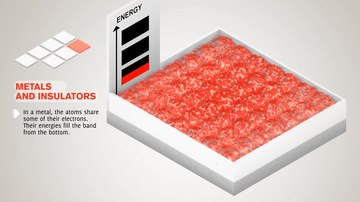
- W półprzewodnikowej lub zespołu izolatora poziom Fermiego jest otoczony przez pasmo wzbronione, zwane pasmo wzbronione (w celu odróżnienia go od innych pasma wzbronione w strukturze pasma). Najbliższy zespół powyżej pasmo wzbronione nazywa się zespół przewodzenia , a najbliżej zespół pod pasmo wzbronione jest nazywana w paśmie walencyjnym . Nazwa „pasmo walencyjne” została ukuta przez analogię do chemii, ponieważ w półprzewodnikach (i izolatorach) pasmo walencyjne jest zbudowane z orbitali walencyjnych .
- W metalu lub semimetalu poziom Fermiego znajduje się wewnątrz jednego lub więcej dozwolonych pasm. W półmetalach pasma są zwykle określane jako „pasmo przewodnictwa” lub „pasmo walencyjne” w zależności od tego, czy transport ładunku jest bardziej podobny do elektronu czy dziury, analogicznie do półprzewodników. Jednak w wielu metalach pasma nie są ani podobne do elektronów, ani dziur, i często nazywane są po prostu „pasmami walencyjnymi”, ponieważ są wykonane z orbitali walencyjnych. Pasma wzbronione w strukturze pasmowej metalu nie są ważne dla fizyki niskich energii, ponieważ są zbyt daleko od poziomu Fermiego.
Teoria w kryształach
Ansatz jest szczególnym przypadkiem fal elektronów w okresowym krystalicznej za pomocą twierdzenia Blocha jako traktowane ogólnie w dynamicznej teorii dyfrakcji . Każdy kryształ jest strukturą periodyczną, którą można scharakteryzować za pomocą sieci Bravais , a dla każdej sieci Bravais możemy wyznaczyć sieć odwrotną , która zawiera okresowość w zestawie trzech odwrotnych wektorów sieci ( b 1 , b 2 , b 3 ). Teraz każdy potencjał okresowy V( r ), który ma taką samą okresowość jak sieć bezpośrednia, może być rozszerzony jako szereg Fouriera, którego jedynymi nieznikającymi składowymi są te związane z odwrotnymi wektorami sieciowymi. Tak więc rozszerzenie można zapisać jako:

gdzie K = m 1 b 1 + m 2 b 2 + m 3 b 3 dla dowolnego zbioru liczb całkowitych (m 1 ,m 2 ,m 3 ).
Na podstawie tej teorii można podjąć próbę przewidzenia struktury pasmowej konkretnego materiału, jednak większość metod ab initio obliczeń struktury elektronowej nie pozwala przewidzieć obserwowanego pasma wzbronionego.
Przybliżenie prawie swobodnego elektronu
W przybliżeniu swobodnych elektronów interakcje między elektronami są całkowicie ignorowane. To przybliżenie pozwala na wykorzystanie twierdzenia Blocha, które mówi, że elektrony w potencjale okresowym mają funkcje falowe i energie, które są okresowe w wektorze falowym aż do stałego przesunięcia fazowego pomiędzy sąsiednimi odwrotnymi wektorami sieciowymi . Konsekwencje okresowości opisuje matematycznie funkcja twierdzenia Blocha:

gdzie funkcja jest okresowa w sieci krystalicznej, czyli

- .

Tutaj indeks n odnosi się do n-tego pasma energii, wektor falowy k jest związany z kierunkiem ruchu elektronu, r jest położeniem w krysztale, a R jest położeniem miejsca atomowego.
Model NFE sprawdza się szczególnie dobrze w materiałach takich jak metale, gdzie odległości między sąsiednimi atomami są niewielkie. W takich materiałach nakładanie się orbitali atomowych i potencjałów na sąsiednie atomy jest stosunkowo duże. W takim przypadku funkcję falową elektronu można aproksymować (zmodyfikowaną) falą płaską. Struktura pasmowa metalu takiego jak aluminium zbliża się nawet do przybliżenia pustej siatki .
Model ciasnego wiązania
Skrajność przeciwna do przybliżenia prawie swobodnego elektronu zakłada, że elektrony w krysztale zachowują się podobnie jak zespół atomów składowych. Ten mocno modelu wiązania przyjmuje rozwiązanie niezależna od czasu pojedynczego elektronu równanie Schrödingera jest także zbliżoną do liniowej kombinacji z orbitali atomowych .


- ,

gdzie współczynniki są dobierane tak, aby dać najlepsze przybliżone rozwiązanie tego formularza. Indeks n odnosi się do poziomu energii atomowej, a R odnosi się do miejsca atomowego. Bardziej dokładne podejście wykorzystujące ten pomysł wykorzystuje funkcje Wanniera , zdefiniowane przez:

- ;

w którym jest okresowa część twierdzenia Blocha, a całka jest nad strefą Brillouina . Tutaj indeks n odnosi się do n-tego pasma energii w krysztale. Funkcje Wanniera są zlokalizowane w pobliżu miejsc atomowych, jak orbitale atomowe, ale zdefiniowane w kategoriach funkcji Blocha są dokładnie powiązane z rozwiązaniami opartymi na potencjale kryształu. Funkcje Wanniera w różnych miejscach atomowych R są ortogonalne. Funkcje Wanniera można wykorzystać do utworzenia rozwiązania Schrödingera dla n-tego pasma energii jako:

- .

Model TB dobrze sprawdza się w materiałach o ograniczonym nakładaniu się orbitali atomowych i potencjałów na sąsiednich atomach. Struktury pasmowe materiałów takich jak Si , GaAs , SiO 2 i diament są dobrze opisane przez TB-Hamiltonian na podstawie orbitali atomowych sp 3 . W metalach przejściowych mieszany model TB-NFE jest używany do opisania szerokiego pasma przewodzenia NFE i wąskich osadzonych pasm d TB. Funkcje radialne atomowej części orbitalnej funkcji Wanniera najłatwiej obliczyć za pomocą metod pseudopotencjalnych . Obliczenia struktury pasmowej NFE, TB lub kombinowanej NFE-TB, czasami rozszerzone o przybliżenia funkcji falowej oparte na metodach pseudopotencjalnych, są często wykorzystywane jako ekonomiczny punkt wyjścia do dalszych obliczeń.
Model KKR
Metoda KKR, zwana także „teorią wielokrotnego rozpraszania” lub metodą funkcji Greena, znajduje stacjonarne wartości odwrotnej macierzy przejść T, a nie hamiltonianu. Implementacja wariacyjna została zasugerowana przez Korringa , Kohna i Rostockera i jest często określana jako metoda Korringa-Kohn-Rostoker . Najważniejszymi cechami sformułowania funkcji KKR lub Greena są: (1) oddziela dwa aspekty problemu: strukturę (położenie atomów) od rozproszenia (tożsamość chemiczna atomów); oraz (2) Funkcje Greena zapewniają naturalne podejście do zlokalizowanego opisu właściwości elektronowych, które można dostosować do stopów i innych nieuporządkowanych układów. Najprostsza forma tego przybliżenia centruje nienakładające się na siebie kule (nazywane puszkami do muffinek ) na pozycjach atomowych. W tych regionach potencjał doświadczany przez elektron jest przybliżony tak, aby był sferycznie symetryczny względem danego jądra. W pozostałym obszarze śródmiąższowym potencjał ekranowany jest aproksymowany jako stała. Wymuszana jest ciągłość potencjału pomiędzy sferami skupionymi wokół atomu a obszarem śródmiąższowym.
Teoria funkcjonału gęstości
W najnowszej literaturze z dziedziny fizyki znaczna większość struktur elektronowych i wykresów pasmowych jest obliczana przy użyciu teorii funkcji gęstości (DFT), która nie jest modelem, ale raczej teorią, tj. mikroskopową teorią pierwszych zasad fizyki materii skondensowanej, która próbuje aby poradzić sobie z problemem wielociałowym elektron-elektron poprzez wprowadzenie terminu wymiany i korelacji w funkcjonale gęstości elektronowej . Pasma obliczone DFT są w wielu przypadkach zgodne z pasmami zmierzonymi eksperymentalnie, na przykład za pomocą kątowej spektroskopii fotoemisyjnej (ARPES). W szczególności kształt pasma jest zazwyczaj dobrze odtworzony przez DFT. Ale są też systematyczne błędy w pasmach DFT w porównaniu z wynikami eksperymentu. W szczególności DFT wydaje się systematycznie zaniżać o około 30-40% przerwę energetyczną w izolatorach i półprzewodnikach.
Powszechnie uważa się, że DFT jest teorią przewidywania stanu podstawowego właściwości systemu tylko (np całkowita energia The struktury atomowej , itd.) I że stanie wzbudzonym właściwości nie można ustalić przez DFT. To błędne przekonanie. W zasadzie DFT może określić dowolną właściwość (stan podstawowy lub stan wzbudzony) układu, mając do dyspozycji funkcjonał, który odwzorowuje gęstość stanu podstawowego na tę właściwość. To jest istota twierdzenia Hohenberga-Kohna. W praktyce jednak nie istnieje żaden znany funkcjonał, który odwzorowuje gęstość stanu podstawowego na energie wzbudzenia elektronów w materiale. Zatem to, co w literaturze jest cytowane jako wykres pasmowy DFT, jest reprezentacją energii DFT Kohna–Shama , tj. energii fikcyjnego układu nieoddziałującego, układu Kohna–Shama, który w ogóle nie ma żadnej fizycznej interpretacji. Struktury elektronowej Kohna-Shama nie należy mylić z rzeczywistą, quasi-cząstkową strukturą elektronową systemu, a dla energii Kohna-Shama nie ma twierdzenia Koopmansa , jak w przypadku energii Hartree-Focka, które można naprawdę uznać za przybliżenie energii quasicząstek . Stąd w zasadzie DFT oparte na Kohn-Sham nie jest teorią pasmową, tj. teorią nadającą się do obliczania pasm i wykresów pasm. W zasadzie do obliczenia rzeczywistej struktury pasmowej można zastosować DFT zależne od czasu, chociaż w praktyce jest to często trudne. Popularnym podejściem jest użycie funkcji hybrydowych , które zawierają część dokładnej wymiany Hartree-Fock; powoduje to znaczną poprawę przewidywanych przerw wzbronionych półprzewodników, ale jest mniej niezawodne w przypadku metali i materiałów o szerokiej przerwie wzbronionej.
Metody funkcji Greena i przybliżenie ab initio GW
Aby obliczyć pasma obejmujące oddziaływanie elektronów z elektronami efekty wielociałowe , można skorzystać z tak zwanych metod funkcji Greena . Rzeczywiście, wiedza o funkcji Greena w systemie dostarcza zarówno podstawy (całkowita energia), jak i obserwabli stanu wzbudzonego systemu. Biegunami funkcji Greena są energie quasicząstek, pasma ciała stałego. Funkcję Greena można obliczyć, rozwiązując równanie Dysona, gdy znana jest energia własna układu. W przypadku rzeczywistych układów, takich jak ciała stałe, energia własna jest bardzo złożoną wielkością i zwykle do rozwiązania problemu potrzebne są przybliżenia. Jednym z takich przybliżeń jest przybliżenie GW , tak zwane z postaci matematycznej, w której energia własna przyjmuje jako iloczyn Σ = GW funkcji Greena G i oddziaływania ekranowanego dynamicznie W . To podejście jest bardziej istotne przy obliczaniu wykresów pasmowych (a także ilości poza nimi, takich jak funkcja spektralna) i może być również sformułowane w sposób całkowicie ab initio . Przybliżenie GW wydaje się zapewniać przerwy wzbronione izolatorów i półprzewodników zgodnie z eksperymentem, a tym samym koryguje systematyczne niedoszacowanie DFT.
Dynamiczna teoria pola średniego
Chociaż przybliżenie prawie swobodnego elektronu jest w stanie opisać wiele właściwości struktur pasmowych elektronów, jedną z konsekwencji tej teorii jest to, że przewiduje ona taką samą liczbę elektronów w każdej komórce elementarnej. Jeśli liczba elektronów jest nieparzysta, spodziewalibyśmy się, że w każdej komórce elementarnej znajduje się niesparowany elektron, a zatem pasmo walencyjne nie jest w pełni zajęte, co czyni materiał przewodnikiem. Jednak materiały takie jak CoO, które mają nieparzystą liczbę elektronów na komórkę elementarną, są izolatorami, co jest w bezpośrednim konflikcie z tym wynikiem. Ten rodzaj materiału jest znany jako izolator Motta i wymaga uwzględnienia szczegółowych oddziaływań elektron-elektron (traktowanych jedynie jako uśredniony wpływ na potencjał kryształu w teorii pasmowej) w celu wyjaśnienia rozbieżności. Model Hubbarda jest przybliżoną teorią, która może obejmować te interakcje. Można ją traktować nieperturbująco w ramach tzw. dynamicznej teorii średniego pola , która stara się wypełnić lukę między przybliżeniem prawie swobodnego elektronu a granicą atomową. Formalnie jednak stany nie oddziałują w tym przypadku, a koncepcja struktury pasmowej nie jest adekwatna do opisania tych przypadków.
Inni
Obliczanie struktur pasmowych jest ważnym tematem w teoretycznej fizyce ciała stałego . Oprócz modeli wymienionych powyżej, inne modele obejmują:
- Aproksymacja pustej sieci : „struktura pasmowa” obszaru wolnej przestrzeni, który został podzielony na sieć.
- Teoria perturbacji k·p jest techniką, która umożliwia przybliżone opisanie struktury pasmowej za pomocą zaledwie kilku parametrów. Technika ta jest powszechnie stosowana w przypadku półprzewodników , a parametry modelu są często określane eksperymentalnie.
- Model Kroniga-Penneya , jednowymiarowy prostokątny model studni przydatny do zilustrowania tworzenia się pasm. Choć jest prosty, przewiduje wiele ważnych zjawisk, ale nie jest ilościowy.
- Model Hubbarda
Strukturę pasmową uogólniono na wektory falowe, które są liczbami zespolonymi , co skutkuje tak zwaną złożoną strukturą pasmową , która jest interesująca na powierzchniach i interfejsach.
Każdy model bardzo dobrze opisuje niektóre typy brył, a inne słabo. Model prawie swobodnych elektronów działa dobrze w przypadku metali, ale słabo w przypadku niemetali. Model ciasnego wiązania jest niezwykle dokładny dla izolatorów jonowych, takich jak sole metalohalogenkowe (np. NaCl ).
Diagramy pasmowe
Aby zrozumieć, jak zmienia się struktura pasmowa w stosunku do poziomu Fermiego w przestrzeni rzeczywistej, wykres struktury pasmowej jest często najpierw upraszczany w postaci diagramu pasmowego . Na schemacie pasmowym oś pionowa jest energią, podczas gdy oś pozioma reprezentuje rzeczywistą przestrzeń. Linie poziome reprezentują poziomy energii, a bloki oznaczają pasma energii. Gdy linie poziome na tym schemacie są pochylone, to energia poziomu lub pasma zmienia się wraz z odległością. Schematycznie przedstawia to obecność pola elektrycznego w układzie krystalicznym. Diagramy pasmowe są przydatne do powiązania ogólnych właściwości struktury pasmowej różnych materiałów ze sobą, gdy są ze sobą stykane.
Zobacz też
- Felix Bloch – pionier w teorii struktury pasmowej
- Alan Herries Wilson – pionier w teorii struktury pasmowej
- Inżynieria przerwy wzbronionej - proces zmiany struktury pasmowej materiału
Bibliografia
Bibliografia
- Charles Kittel (1996). Wprowadzenie do Fizyki Ciała Stałego (wyd. siódme). Nowy Jork: Wiley. Numer ISBN 978-0-471-11181-8.
Dalsza lektura
- Mikroelektronika , Jacob Millman i Arvin Gabriel, ISBN 0-07-463736-3 , Tata McGraw-Hill Edition.
- Fizyka ciała stałego , Neil Ashcroft i N. David Mermin, ISBN 0-03-083993-9
- Elementary Solid State Physics: Principles and Applications , M. Ali Omar, ISBN 0-201-60733-6
- Elektroniczne i optoelektroniczne właściwości struktur półprzewodnikowych - rozdział 2 i 3 autorstwa Jasprit Singh, ISBN 0-521-82379-X
- Struktura elektronowa: podstawowa teoria i metody praktyczne autorstwa Richarda Martina, ISBN 0-521-78285-6
- Fizyka materii skondensowanej Michael P. Marder, ISBN 0-471-17779-2
- Metody obliczeniowe w fizyce ciała stałego VV Nemoshkalenko i NV Antonov, ISBN 90-5699-094-2
- Elementarna struktura elektroniczna Walter A. Harrison, ISBN 981-238-708-0
- Pseudopotencjały w teorii metali Walter A. Harrison, WA Benjamin (Nowy Jork) 1966
- Tutorial on Bandstructure Methods autorstwa dr Vasileski (2008)
Zewnętrzne linki
-
 Multimedia związane z elektronicznymi strukturami pasm w Wikimedia Commons
Multimedia związane z elektronicznymi strukturami pasm w Wikimedia Commons - Animacja, zastosowania i badania dotyczące fizyki kwantowej i teorii pasmowej (Université Paris Sud)