
Kąt Bürgi-Dunitz - Bürgi–Dunitz angle



Kąt Bürgi-Dunitz (kąt BD) jest jedną z dwóch kątów, który w pełni określają geometrię „atak” (podejście poprzez zderzenia) z nukleofilem, na trójkątny nienasyconego centrum w cząsteczce , pierwotnie karbonylo środkowej w organicznych ketonu , ale obecnie rozciąga się na karbonylki aldehydów , estrów i amidów , a także na alkeny (olefiny).
Praktycznie rzecz biorąc, kąty Bürgi-Dunitz i Flippin-Lodge były kluczowe dla rozwoju zrozumienia chiralnej syntezy chemicznej , a konkretnie zjawiska asymetrycznej indukcji podczas ataku nukleofilowego w zaburzonych centrach karbonylowych (patrz modele Cram-Felkin-Anh i Nguyen ).
Ponadto zasady stereoelektroniczne, które leżą u podstaw nukleofili przyjmujących określony zakres kątów Bürgi-Dunitz, mogą przyczyniać się do stabilności konformacyjnej białek i są przywoływane w celu wyjaśnienia stabilności poszczególnych konformacji cząsteczek w jednej z hipotez o chemicznym pochodzeniu życia .
Wstęp
Dokładniej, w przypadku ataku nukleofilowego na karbonyl, kąt Bürgi-Dunitz jest zdefiniowany jako kąt wiązania Nu-CO, gdzie Nu jest tu używane do identyfikacji atomu nukleofila, który tworzy wiązanie z atomem węgla, C. Kąt został nazwany na cześć krystalografów Hansa-Beata Bürgi i Jacka D. Dunitza , jego pierwszych starszych badaczy.
Kąt BD przyjęty podczas podejścia nukleofila do trygonalnego nienasyconego elektrofila zależy przede wszystkim od kształtu orbitali molekularnych (MO) i zajętości centrum nienasyconego węgla (np. centrum karbonylowego), a tylko wtórnie od orbitali molekularnych nukleofila.
Z dwóch kątów, które definiują geometrię nukleofilowego „ataku”, drugi opisuje „przesunięcie” podejścia nukleofila w kierunku jednego z dwóch podstawników przyłączonych do węgla karbonylowego lub innego centrum elektrofilowego i został nazwany Flippin-Lodge (FL ) autorstwa Claytona Heathcocka po jego współpracownikach Lee A. Flippin i Eric P. Lodge.
Kąty te są ogólnie rozumiane jako kąt zmierzony lub obliczony dla danego systemu, a nie historycznie obserwowany zakres wartości dla oryginalnych aminoketonów Bürgi-Dunitz lub wyidealizowana wartość obliczona dla konkretnego systemu (np. addycja wodorków do formaldehydu ). po lewej). Oznacza to, że kąty BD i FL układu wodorkowo-formadehydowego dają daną parę wartości, podczas gdy kąty obserwowane dla innych układów mogą się różnić w stosunku do tego najprostszego układu chemicznego.
Jako eksperymentalny obserwowalny
Pomiar
Pierwotne pomiary Bürgi-Dunitz dotyczyły serii wewnątrzcząsteczkowych oddziaływań amina - keton karbonylowych w kryształach związków posiadających obie funkcjonalności, np. metadonu i protopiny . Dało to wąski zakres wartości kąta BD (105 ± 5°); odpowiednie obliczenia — obliczenia orbitali molekularnych typu SCF-LCAO — opisujące podejście orbitalu s anionu wodorkowego (H − ) do układu pi najprostszego aldehydu, formaldehydu (H 2 C=O), dały wartość kąta BD 107°.
Stąd Bürgi, Dunitz i wielu innych zauważyli, że pomiary krystalograficzne aminoketonów i oszacowanie obliczeniowe dla najprostszego układu nukleofil-elektrofil były dość bliskie ideału teoretycznego, kąta czworościanu (kąty wewnętrzne czworościanu 109,5°). , a więc zgodne z geometrią uważaną za ważną dla rozwoju stanów przejściowych w atakach nukleofilowych w centrach trygonalnych.
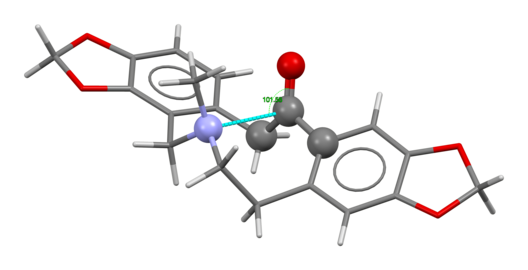
Oddziaływanie amina-karbonyl n→π* w protopinie przy niezwykle krótkiej odległości N···C wynoszącej 2,555 Å i kącie Bürgi-Dunitz 102°.



W strukturze L- metadonu (powyżej, po lewej) zwróć uwagę na trzeciorzędową aminę wystającą w prawym dolnym rogu i grupę karbonylową (C=O) w środku, które angażują się w interakcję wewnątrzcząsteczkową w strukturze krystalicznej (po obrocie wokół łączące je pojedyncze wiązania podczas procesu krystalizacji ).
Podobnie w strukturze protopiny (powyżej, w środku) zwróć uwagę na trzeciorzędową aminę w centrum cząsteczki, część dziesięcioczłonowego pierścienia, oraz grupę C=O naprzeciw niej na pierścieniu; wchodzą one w interakcję wewnątrzcząsteczkową, na którą pozwalają zmiany kątów torsyjnych atomów pierścienia.
Wyjaśnienie teoretyczne


(Po prawej) Rysunek przedstawiający podejście do najwyżej zajętego orbitalu molekularnego typu p (HOMO) nukleofila, takiego jak jon chlorkowy (zielona kula) i najniższego niezajętego orbitalu molekularnego (LUMO) centrum trygonalnego elektrofilowego karbonylku formaldehydu (czarna kula węgiel, czerwona kula tlen, białe kulki wodór). Widok jest prawie z boku, a rozwijające się zniekształcenie poza płaszczyzną karbonylowego atomu węgla jest pominięte dla uproszczenia.
HOMO-LUMO nakładają się
Zbieżność obserwowanych kątów BD może być postrzegana jako wynikająca z potrzeby maksymalizacji nakładania się pomiędzy najwyższym zajętym orbitalem molekularnym ( HOMO ) nukleofila i najniższym niezajętym orbitalem molekularnym ( LUMO ) nienasyconego, trygonalnego centrum elektrofila. (Patrz, dla porównania, powiązana koncepcja chemii nieorganicznej modelu nakładania się kątów).
W przypadku dodania do grupy karbonylowej Homo często typu p orbitalne (na przykład na aminy azotu lub halogenku anionu ) i LUMO jest ogólnie rozumiane jako antibonding π * cząsteczkowej orbitalnej prostopadłej do płaszczyzny zawierającej wiązanie ketonowe C=O i jego podstawniki (patrz rysunek po prawej powyżej). Uważa się, że kąt BD obserwowany dla ataku nukleofilowego zbliża się do kąta, który zapewni optymalne nakładanie się HOMO i LUMO (w oparciu o zasadę obniżania powstałych nowych energii orbitali molekularnych po takim zmieszaniu orbitali o podobnej energii i symetrii z uczestniczących reagentów ). Jednocześnie nukleofil unika nakładania się z innymi orbitalami grupy elektrofilowej, które są niekorzystne dla tworzenia wiązań (niewidoczne na obrazku po prawej, powyżej, ze względu na prostotę R=R'=H w formaldehydzie).
Komplikacje
Oddziaływania elektrostatyczne i Van der Waalsa
Aby zrozumieć przypadki rzeczywistych reakcji chemicznych, skoncentrowany na HOMO-LUMO pogląd został zmodyfikowany poprzez zrozumienie dalszych złożonych, specyficznych dla elektrofilów odpychających i atrakcyjnych oddziaływań elektrostatycznych i Van der Waalsa, które zmieniają kąt wysokościowy BD i odchylają azymutalny kąt Flippin-Lodge w kierunku jednego lub drugiego podstawnika (patrz rysunek powyżej).
Dynamika liniowa i obrotowa
Teoria kąta BD została opracowana na podstawie „zamrożonych” oddziaływań w kryształach, gdzie wpływy dynamiki w układzie (np. łatwo zmieniające się kąty skręcania ) mogą być pomijalne. Jednak większość chemii będącej przedmiotem ogólnego zainteresowania i zastosowania odbywa się poprzez zderzenia cząsteczek opadających w roztworze ; odpowiednio w takich przypadkach uwzględnia się dynamikę.
Ograniczone środowiska w enzymach i nanomateriałach
Co więcej, w ograniczonych środowiskach reakcji, takich jak miejsca wiązania enzymów i nanomateriałów, wczesne dowody sugerują, że kąty BD dla reaktywności mogą być całkiem różne, ponieważ koncepcje reaktywności zakładające nakładanie się orbit podczas losowej kolizji nie mają bezpośredniego zastosowania.
Na przykład, wartość BD wyznaczona dla enzymatycznego rozszczepienia amidu przez proteazę serynową ( subtylizynę ) wynosiła 88°, zupełnie odmienna od wartości wodorkowo-formaldehydowej wynoszącej 107°; ponadto, kompilacja wartości krystalograficznych kąta BD z literatury dla tej samej reakcji, w której pośredniczą różne katalizatory białkowe skupione w 89 ± 7° (tj. tylko nieznacznie przesunięte względem bezpośrednio powyżej lub poniżej węgla karbonylowego). W tym samym czasie wartość FL subtylizyny wynosiła 8°, a wartości kąta FL ze starannego zestawienia skupiły się na 4 ± 6° (tj. tylko nieznacznie przesunięte względem karbonylku; patrz artykuł dotyczący kąta Flippina-Lodge'a ).